目次

骨形成不全症II型(OI Type II)は、COL1A1またはCOL1A2遺伝子のミスセンス変異が引き起こす「優性阻害効果」によって、I型コラーゲンの三重らせん構造が根本から崩壊する最重症の遺伝性骨疾患です。胎内での多発骨折・頭蓋骨石灰化不全・致死的な肺低形成という三重の病態が組み合わさり、出生直後の呼吸不全によって数時間〜数週間以内に死に至る「周産期致死型」と定義されています。
Q. 骨形成不全症II型とはどのような疾患ですか?まず結論だけ知りたいです
A. COL1A1またはCOL1A2遺伝子のミスセンス変異によってI型コラーゲンの構造が崩壊し、優性阻害効果が生じる最重症の遺伝性骨疾患です。胎内多発骨折・極端な肺低形成・頭蓋骨石灰化不全を特徴とし、出生直後の呼吸不全で周産期に死に至るため「周産期致死型」と呼ばれます。ほぼ全例が両親に変異のない新生突然変異(de novo変異)によって散発的に発生します。
- ➤疾患の定義 → Sillence分類の中で最重症、90〜95%がCOL1A1/COL1A2優性変異に起因
- ➤分子メカニズム → Gly-X-Y反復配列のグリシン置換→三重らせん形成障害→優性阻害効果
- ➤主な症状 → 多発骨折・肺低形成・頭蓋石灰化不全・青色強膜・短縮四肢
- ➤鑑別診断 → 低ホスファターゼ症(治療可能)・軟骨無発生症・タナトフォリック骨異形成症との違いを詳解
- ➤診断・検査 → 妊娠中期超音波3基準・X線サブタイプ(IIA/IIB/IIC)・NGS段階的アプローチ
1. 骨形成不全症II型とは:疾患の定義と分類上の位置づけ
骨形成不全症(Osteogenesis Imperfecta:OI)は、I型コラーゲンの合成量低下または分子構造の異常を根本原因とする遺伝性結合組織疾患の総称です。「脆い骨(brittle bone disease)」とも呼ばれ、軽微な外力でも骨折を繰り返す極度の易骨折性を最大の特徴とします。その表現型(症状の現れ方)は非常に幅広く、生涯で数回の骨折を経験する程度の軽症から、胎生期より全身に多発骨折を来し出生後まもなく死亡する最重症まで、連続したスペクトラムを形成しています。
💡 用語解説:Sillence分類とは
1979年にオーストラリアの遺伝学者David Sillenceらが提唱した骨形成不全症の臨床分類です。症状の重症度・遺伝形式・X線所見に基づき、I型(最軽症・非変形性)→ II型(最重症・周産期致死)→ III型(重症変形性)→ IV型(中等症)の4つに大別します。現在では分子遺伝学の進歩により少なくとも19の病型が認識されていますが、臨床診断の枠組みとして今もこの分類が中核をなしています。
この分類の中で、骨形成不全症II型(OI Type II)は「周産期致死型(Perinatally lethal OI)」として位置づけられ、全OIサブタイプの中で最も重篤なフェノタイプを示します。I型が軽症の対極にあるのに対し、II型は胎児発生の段階から骨格系の構造的完全性に致命的な破綻を来します。臨床的には90〜95%の症例がCOL1A1またはCOL1A2遺伝子の優性変異に起因し、ほぼ全例が新生突然変異(de novo変異)によって散発的に発生します。
2023年に発表された遺伝性骨格疾患の最新疾病分類(Nosology 2023 revision)では、疾患を「表現型と責任遺伝子の組み合わせ」で定義する「ダイアディック・アプローチ」が正式に採用されました。この枠組みに従えば、OI II型も「COL1A1関連骨形成不全症(周産期致死型)」あるいは「COL1A2関連骨形成不全症(周産期致死型)」として、より精緻に定義されるようになっています。
💡 用語解説:周産期致死(しゅうさんきちし)とは
「周産期」とは出産前後の時期(概ね妊娠22週以降〜生後1週間)を指します。「周産期致死」とは、この時期に生命を失う、すなわち死産または生後まもない新生児期の死亡を意味します。OI II型の場合、出生時に骨格・胸郭・肺の発達が致命的に障害されているため、人工呼吸管理を行っても多くの場合、生後数時間〜数週間以内に呼吸不全で死亡します。現時点では根治的治療法は存在しません。

2. 原因遺伝子と分子病態メカニズム
骨形成不全症II型の病態を理解するには、I型コラーゲンという巨大タンパク質の構造と、それをコードする遺伝子の変異がなぜ致死的な結果をもたらすのかを、分子レベルから理解することが重要です。
💡 用語解説:I型コラーゲンとは
I型コラーゲンは、骨・皮膚・腱・靭帯・象牙質の主要構成成分である細胞外マトリックスタンパク質です。2本のα1(I)鎖と1本のα2(I)鎖が互いに強固に右巻きに編み込まれた三重らせん構造(ヘテロ三量体)を形成します。この三重らせんを維持するためには、鎖のアミノ酸配列に「Gly-X-Y」という繰り返し構造が連続していることが絶対条件です。グリシン(Gly)は最も小さなアミノ酸であり、三重らせんの狭い中心軸に収まることができる唯一のアミノ酸です。
原因遺伝子:COL1A1とCOL1A2
I型コラーゲンをコードするのが、染色体17q21.33に位置するCOL1A1遺伝子と、染色体7q21.3に位置するCOL1A2遺伝子です。臨床的に骨形成不全症と診断された患者の実に90〜95%は、これらいずれかの遺伝子にヘテロ接合性の病的バリアントを持つ常染色体優性遺伝形式の疾患です。
💡 用語解説:常染色体優性遺伝
「常染色体」とは性染色体(X・Y)以外の染色体のこと。「優性(顕性)」とは、2本の染色体のうちどちらか1本に変異があるだけで症状が現れる遺伝形式です。OI II型は常染色体優性遺伝ですが、患者自身は致死性ゆえに生殖年齢に達して変異を伝達することがないため、実質的に全症例が両親に変異のない「新生突然変異(de novo変異)」として発生します。
なぜ致死的になるのか:優性阻害効果のメカニズム
同じCOL1A1/COL1A2遺伝子の変異でも、I型OI(軽症)とII型OI(致死)ではまったく異なる病態メカニズムが働いています。
軽症のI型OIでは、多くの場合COL1A1遺伝子の「ヌル変異(ナンセンス変異・フレームシフト変異)」によって変異アレルからのα1鎖産生が完全に停止します。産生されるI型コラーゲンの総量は半分に減りますが、できあがったコラーゲン分子の構造自体は正常です(量的異常)。これをハプロ不全(haploinsufficiency)といいます。
💡 用語解説:優性阻害効果(ドミナントネガティブ効果)
OI II型の多くは、Gly-X-Y反復配列の中のグリシン残基が別のアミノ酸(アルギニン・セリン・バリンなど)に置き換わるミスセンス変異によって引き起こされます。このかさばったアミノ酸が三重らせんの中心軸への「詰まり」を生じさせ、らせん形成が途中で物理的に阻害されます。I型コラーゲンは3本の鎖から成るため、変異アレルの産生物が混入した三重らせんは全体の約75%に及び、正常分子を圧倒します。このように異常タンパク質が正常タンパク質の機能まで構造的に破綻させる現象を「優性阻害効果(ドミナントネガティブ効果)」と呼びます。ハプロ不全よりはるかに重篤な病態を引き起こします。
構造異常を来した変異コラーゲンの大部分は小胞体内で不良タンパク質として認識・分解され、骨芽細胞に深刻な小胞体ストレス(ER stress)を引き起こします。さらに細胞外に分泌された一部の異常コラーゲンが正常なコラーゲン線維(フィブリル)の自己組織化を阻害し、石灰化の足場となる骨基質の完全性を根本から破壊します。大規模な患者データ解析からは、COL1A1鎖での変異がCOL1A2鎖での変異よりも致死的フェノタイプ(II型)となるリスクが有意に高いことが判明しています。
💡 用語解説:過剰修飾(overmodification)
ミスセンス変異によってグリシン置換が生じ、三重らせん形成がその部位で停滞すると、変異部位よりN末端側の鎖が通常よりも長時間にわたって修飾酵素(プロリル水酸化酵素・リシル水酸化酵素など)にさらされ続けます。その結果、特定領域で正常状態を超える水酸化・糖鎖付加(過剰修飾)が生じます。この分子量増大を利用してSDS-ポリアクリルアミドゲル電気泳動(SDS-PAGE)上で「泳動速度の遅延」として検出するのが生化学的コラーゲン分析の原理です。
💡 用語解説:新生突然変異(de novo変異)と生殖細胞系モザイク
de novo変異とは、両親の体細胞には存在せず、精子・卵子の形成時または受精後に新たに生じた変異で、OI II型のほぼ全例がこれに該当します。一方、生殖細胞系モザイク(germline mosaicism)とは、外見上健常な親の精子または卵子を産生する細胞の一部に変異が潜んでいる状態です。この場合、次の妊娠でもOI II型が再発するリスクが存在します。頻度は低いですが、遺伝カウンセリングで必ず説明が必要な重要な概念です。
3. 主な症状と全身への影響
骨形成不全症II型の異常は、軽症例のように生後数ヶ月以降に初めて骨折を経験するのとは根本的に異なり、胎生期からすでに全身の骨格系に致命的な破綻が生じています。発生の極めて初期段階から骨格のモデリングが阻害されるため、影響は骨格のみならず結合組織全体に及びます。
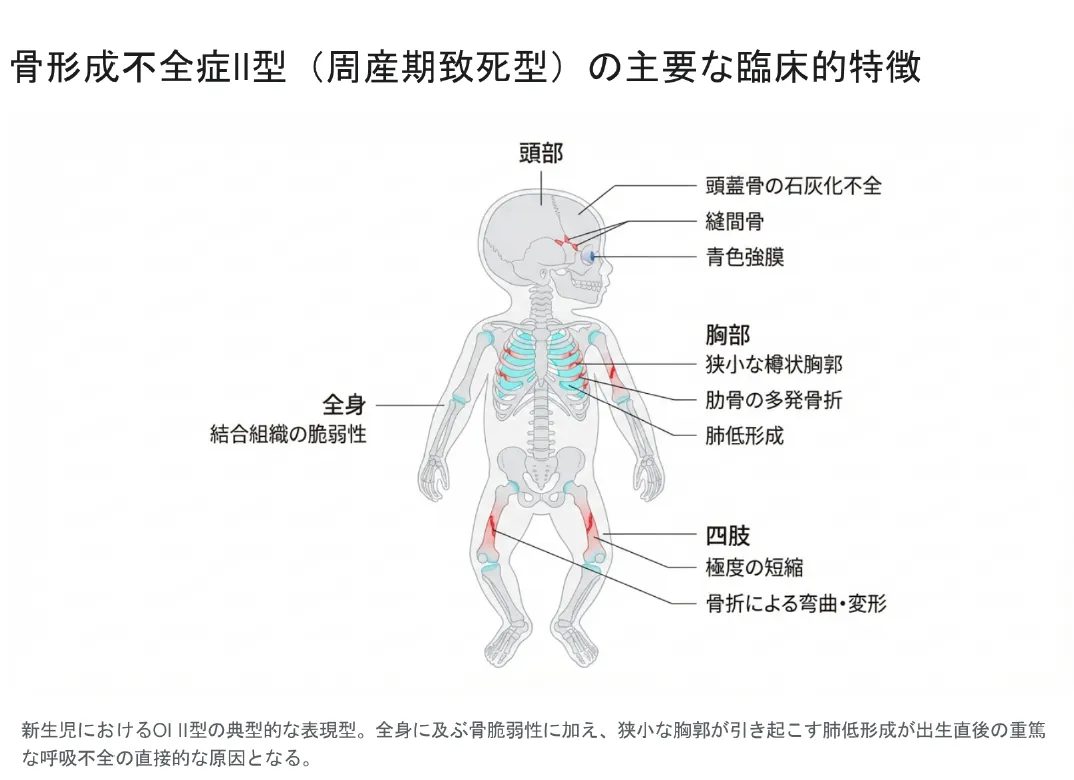
新生児におけるOI II型の典型的な表現型。全身に及ぶ骨脆弱性に加え、狭小な胸郭が引き起こす肺低形成が出生直後の重篤な呼吸不全の直接的な原因となる。
🦴 骨格系(全身)
- 胎内での多発性骨折(長管骨・肋骨)
- 長管骨の著明な短縮・弯曲・ひしゃげ
- 骨皮質の菲薄化・広範な骨減少症
- 修復過程を示す仮骨形成の痕跡
💨 胸郭・呼吸器(直接死因)
- 極端に狭小な胸郭(樽状胸郭)
- 肋骨の多発性骨折
- 致死的な肺低形成
- 出生直後からの新生児呼吸窮迫
🧠 頭蓋・顔面
- 重度の頭蓋骨化不全(薄く柔らかい頭蓋冠)
- 多数の縫間骨(Wormian bones)
- 相対的大頭症・「兜状」頭蓋
- 逆三角形の顔立ち(前額突出・小さい下顎)
👁️ その他の結合組織
- 青色強膜(ほぼ全例)
- 皮膚菲薄化・皮下出血しやすい
- 関節の過弛緩または拘縮
- 全身の筋力低下
💡 用語解説:肺低形成(pulmonary hypoplasia)とは
肺が正常な大きさ・構造に発育しない状態です。OI II型では、極端に狭小化・構造的支持を失った胸郭が、胎児期の肺の正常な拡張と実質組織の発育を著しく妨げます。その結果、出生直後より直ちに重篤な呼吸窮迫を呈し、人工呼吸管理を行っても多くは生後数時間〜数週間以内に死亡します。これがOI II型の直接的な死因の大部分を占めます。
💡 用語解説:青色強膜(blue sclerae)とは
眼球の「白目」の部分(強膜)が青色または濃い灰色を呈する所見です。強膜の結合組織を構成するI型コラーゲン線維網が異常に薄く形成不全であるため、下層のブドウ膜(uvea)のメラニン色素が物理的に透けて見えることで生じます。OI II型ではほぼ全例で認められる特徴的所見です。
💡 用語解説:縫間骨(Wormian bones)とは
頭蓋骨の縫合部(骨と骨の接合部)に多数出現する小さな余分な骨のことです。頭蓋骨の骨化遅延・骨形成不全の結果として現れ、X線検査で特徴的に認められます。OI II型に特有の所見ではありませんが、重度の頭蓋骨化不全と組み合わさって認められることがほとんどです。
4. 鑑別診断:超音波で似て見える疾患との違い
OI II型は妊娠中期の胎児超音波で発見されることが大半ですが、超音波・X線像が酷似する別疾患が複数存在します。なかでも先天性低ホスファターゼ症は、適切な治療を行えば予後が劇的に改善する疾患であるため、OI II型との鑑別は単なる学術的な問題ではなく、児の生命予後に直結する臨床上の最重要課題です。
⚠️ 先天性低ホスファターゼ症(Congenital Hypophosphatasia)
原因:ALPL遺伝子変異。全身の重篤な石灰化不全を呈し、超音波・X線像がOI II型に極めて酷似する。
鑑別の決め手:血中アルカリホスファターゼ(ALP)値の著明な低下、ALPL遺伝子検査。
✅ 酵素補充療法(アスホターゼ アルファ)による劇的な生命予後改善が報告されており、確実な鑑別が治療選択に直結します。
軟骨無発生症(Achondrogenesis)
共通点:OI II型同様、重度の四肢短縮と骨化不全を認める。
鑑別のポイント:多発骨折や仮骨形成は軟骨無発生症の主要画像所見ではない。原因遺伝子も異なる(COL2A1など)。
タナトフォリック骨異形成症(Thanatophoric Dysplasia)
共通点:FGFR3遺伝子変異によるOI II型と同様に致死的な骨格異形成。極端な長管骨短縮・狭小胸郭を呈する。
鑑別のポイント:大腿骨が電話の受話器状に弯曲し、頭蓋骨の石灰化は比較的保たれ、骨折は見られない。
💡 用語解説:先天性低ホスファターゼ症(Hypophosphatasia)
ALPL遺伝子の変異によって、骨・歯の石灰化に不可欠な酵素「組織非特異型アルカリホスファターゼ(TNSALP)」が欠乏する遺伝性疾患です。周産期重症型はOI II型と酷似した骨化不全・多発骨折を呈しますが、血中ALP値の著明な低下が特徴的な生化学的所見です。近年、酵素補充療法(商品名:ストレンジック)によって致死性から生存可能へと予後が劇的に変化した疾患であり、OI II型との鑑別は治療可否を決定する緊急課題です。
5. 診断基準と遺伝子検査の進め方
胎児期超音波診断:3つの決定的基準
OI II型の致死性から、妊娠中期(特に20週未満)の定期胎児超音波スクリーニングで発見されることが大半です。超音波精度は通常、妊娠16〜24週の間に高まります。OI再発リスクのある家系では、以下の3つの超音波診断基準を全て満たす場合、II型の確実な診断に結びつくとされています。
OI II型を確実に診断する超音波3基準
- 基準①多発性の骨折(Multiple fractures):大腿骨・脛骨・上腕骨などの長管骨に異常な屈曲、骨折に伴う仮骨形成が描出される
- 基準②頭蓋冠の石灰化低下(Demineralization of the calvaria):超音波の透過性が異常に亢進し、プローブで押すと頭蓋骨がゴムまりのように凹む(compressibility)
- 基準③大腿骨長の著明な短縮(Severe femoral shortening):在胎週数の平均値から3標準偏差(SD)以上下回る
なお、妊娠17週以降の超音波所見が完全に正常であれば、致死性のII型は除外できるとされています。ただし、20週未満の長管骨正常所見は軽症型OIを完全には排除しません。
X線像に基づく放射線学的サブタイプ分類(IIA・IIB・IIC)
OI II型は、四肢の長管骨と胸郭肋骨のX線形態の差異に基づいて、さらに3つのサブタイプ(IIA・IIB・IIC)に分類されます。これは骨基質異常に対する組織の反応性の差を反映しています。
| サブタイプ | 長管骨の特徴 | 肋骨の特徴 |
|---|---|---|
| IIA型 (最多) |
著しいモデリング不全。短く・幅広く・ひしゃげた(crumpled)変形 | 短く幅広い。連続的な数珠状変形(continuous beading)が病理特異的所見。最大の特徴 |
| IIB型 | IIA型とほぼ同様。短く・幅広く・ひしゃげた変形 | 正常〜軽度菲薄化。連続的数珠状変形は欠如または最小限・不連続 |
| IIC型 | IIA/IIBよりやや細く長いが、モデリング不全は存在 | 極端に細い肋骨+数珠状変形を伴う |
いずれのサブタイプにも共通する所見:頭蓋冠の重度骨化欠損・縫間骨・脊椎における扁平椎(platyspondyly)
💡 用語解説:扁平椎(platyspondyly)とは
椎体(背骨を構成する個々の骨)が扁平化している状態です。OI II型では、脊椎の骨格モデリング不全によってほぼ全例に認められる特徴的なX線所見です。重度の場合、脊椎の支持機能が著しく損なわれます。
段階的(Tiered)分子遺伝学的診断アルゴリズム
現在、OIの確定診断における第一選択は次世代シーケンシング(NGS)を用いた遺伝子解析です。95%以上がCOL1A1/COL1A2変異に起因するため、費用対効果の高い段階的アプローチが推奨されています。
骨形成不全症における段階的診断アルゴリズム
全コーディングエクソン+隣接スプライス領域を20X以上で解析。サンガー法による確認も実施。→ 陽性の場合:優性OI確定
高密度ターゲットオリゴアレイ等でコーディング配列・上流領域の大きなゲノム構造異常を探索。→ 陽性の場合:優性OI確定
稀な常染色体劣性OI(CRTAP・LEPRE1/P3H1等)や他の遺伝性骨疾患を網羅的に探索。→ 陽性の場合:劣性OI等確定
SDS-尿素-PAGEによる過剰修飾(電気泳動遅延)の検出。感度約90%。点突然変異が特定できない症例の補完的診断ツール。
出生前診断では末梢血・唾液・培養羊水細胞・絨毛採取(CVS)から抽出したDNAを使用。羊水細胞はI型コラーゲンを十分合成しないため生化学的分析には不適(遺伝子解析のみに使用)。
💡 用語解説:次世代シーケンシング(NGS)
従来のサンガー法(1遺伝子ずつ解析)と異なり、数百〜数万の遺伝子を同時に・高速で・高精度に解析できる最新のDNA配列解析技術の総称です。ターゲット遺伝子パネル・全エクソームシーケンス(WES)・全ゲノムシーケンス(WGS)などが含まれます。OIの診断では、まずCOL1A1/COL1A2のターゲットNGSから始め、陰性の場合に段階的に広い解析へと移行します。全てのバリアントはACMG/AMPガイドラインに基づいて病原性評価されます。
6. 治療と長期管理:緩和ケアの位置づけ
骨形成不全症II型は「周産期致死型」の定義通り、現時点において根本的な病態を覆し得る根治的治療法は存在せず、予後は極めて不良です。したがって、臨床マネジメントの目標は寿命延長や機能回復ではなく、苦痛の緩和(Palliative care)と生命維持に向けた支持的ケアに限定されます。このプロセスには、産婦人科医・新生児科医・臨床遺伝専門医・医療ソーシャルワーカーからなる集学的チームの密接な連携が不可欠です。
OI II型に対する現実的な対応
- 出生後の苦痛緩和(緩和ケア)
- 人工呼吸管理(一時的支持)
- 家族への心理的サポート
- 多職種チームによる意思決定支援
非致死性OI(III・IV型)では有効な治療
- ビスホスホネート製剤静脈内投与(骨密度維持・骨折頻度低減)
- 髄内釘(ロッド)挿入による整形外科的介入
- 理学療法・作業療法
- 成長ホルモン療法(一部の症例)
II型患児では、出生直後からの重篤な肺低形成と胸郭虚脱に起因する呼吸不全が急速に進行するため、ビスホスホネート治療による長期的骨量増加効果を期待する時間的猶予がなく、骨格への外科的再建術が適応となる局面も事実上ありません。
ただし、最も重要な「治療的介入」は正確な鑑別診断の確立です。特に表現型が酷似する先天性低ホスファターゼ症の周産期重症型については、近年の酵素補充療法(アスホターゼ アルファ)により劇的な予後改善が報告されており、OI II型との迅速な鑑別が文字通り「命の分かれ目」となります。遺伝子パネル検査と血中ALP測定を組み合わせた早期鑑別が現代の新生児・胎児医療において最も高い臨床的意義を持ちます。
7. 遺伝カウンセリングの重要性
OI II型の確定診断後(出生前・出生後どちらでも)、臨床遺伝専門医による包括的な遺伝カウンセリングが不可欠です。遺伝カウンセリングで扱われる主な内容は以下の通りです。
- ➤再発リスクの説明:ほぼ全例がde novo変異であり、両親への遺伝は認められません。ただし常染色体優性遺伝のため、患者本人が生存・生殖した場合の子どもへの遺伝確率は理論上50%です。また、生殖細胞系モザイクが存在する場合、次回妊娠でのOI II型再発リスクが上乗せされます(通常より高いが数%程度と推定)。
- ➤次子への出生前診断の選択肢:以前にOI II型の児を出産した既往がある家系では、次の妊娠において絨毛検査(CVS)または羊水検査による出生前遺伝子診断が選択肢となります。特定の変異が同定されていれば確実な診断が可能です。
- ➤予後と意思決定支援:OI II型が確実と判断された場合の周産期管理(帝王切開の適否・分娩場所の選択・出生後の緩和ケア方針など)について、家族の価値観を尊重しながら丁寧に話し合うプロセスが遺伝カウンセリングの中核をなします。
- ➤モザイクリスクの評価:両親の血液・皮膚等からの生殖細胞系モザイク検索は感度に限界がありますが、次子の再発リスク評価のために実施が推奨される場合があります。
8. よくある誤解
誤解①「OIは全て骨折しやすいだけの病気」
OIは広範な結合組織疾患であり、II型では骨折以上に肺低形成による呼吸不全が直接の死因です。「骨が折れやすい」という認識では重症度の実態を捉えられません。
誤解②「親が健康なら遺伝病ではない」
OI II型のほぼ全例はde novo(新生)変異です。両親が完全に健康でも発生するため、「遺伝病は親が罹患していないと起きない」という誤解が診断の遅れを招くことがあります。
誤解③「超音波でOI II型と確定できる」
超音波は強力なスクリーニングツールですが、低ホスファターゼ症など超音波像が酷似する疾患の除外には生化学的検査・遺伝子検査による確定が不可欠です。超音波のみで「OI II型確定」とするのは危険です。
誤解④「次の妊娠では再発しない」
多くはde novo変異のため再発リスクは低いですが、生殖細胞系モザイクが存在する場合は次子に再発する可能性があります。「一度限り」と断言することはできず、次妊娠では必ず遺伝カウンセリングを受けることが重要です。
誤解⑤「II型と診断されたら何もできない」
根治的治療はありませんが、正確な鑑別診断によって類似疾患(低ホスファターゼ症)の治療機会を逃さないこと、そして適切な緩和ケアと家族支援の提供は「できること」です。診断の確実性が家族の選択を支えます。
誤解⑥「OI II型はOI I型の重症版」
I型(ハプロ不全:量的異常)とII型(優性阻害:構造的異常)は分子メカニズムが根本的に異なります。「重い・軽い」の連続体ではなく、質的に異なる病態です。
9. 臨床遺伝専門医からのメッセージ
よくある質問(FAQ)
🏥 出生前診断・遺伝カウンセリングについて
骨形成不全症をはじめとする遺伝性骨疾患・出生前診断に関するご相談は、
臨床遺伝専門医が在籍するミネルバクリニックへお気軽にご相談ください。
関連記事
参考文献
- [1] Osteogenesis Imperfecta – StatPearls – NCBI Bookshelf. [NCBI Bookshelf]
- [2] Osteogenesis imperfecta – Genetics – MedlinePlus. [MedlinePlus]
- [3] Genetics of Osteogenesis Imperfecta – Medscape. [Medscape]
- [4] COL1A1- and COL1A2-Related Osteogenesis Imperfecta – GeneReviews – NCBI Bookshelf. [GeneReviews]
- [5] Radiographic features of osteogenesis imperfecta – PMC. [PMC3731461]
- [6] Perinatal lethal type II osteogenesis imperfecta: a case report – PMC. [PMC4561136]
- [7] Imaging in osteogenesis imperfecta – Hrcak (Paediatr Croat). [Hrcak]
- [8] Osteogenesis Imperfecta Type II: Prenatal Sonographic Diagnosis – PubMed. [PubMed]
- [9] Osteogenesis Imperfecta Test Guide – Laboratory for Precision Medicine, University of Washington. [UW Collagen Diagnostic Lab]
- [10] Biochemical screening of type I collagen in osteogenesis imperfecta – PMC. [PMC2564593]
- [11] Nosology of genetic skeletal disorders: 2023 revision – PMC. [PMC10081954]
- [12] Dissecting the phenotypic variability of osteogenesis imperfecta – Disease Models & Mechanisms. [DMM]
- [13] Osteogenesis imperfecta – TheFetus.net. [TheFetus.net]
- [14] Osteogenesis Imperfecta type II – Clinical Genetic Test – GTR – NCBI. [NCBI GTR]
- [15] About Osteogenesis Imperfecta – Genome.gov. [Genome.gov]






