目次

Bohring-Opitz症候群(BOS)は、第20染色体上のASXL1遺伝子に生じる新生(de novo)トランケーティング変異によって引き起こされる、100万人に1人未満という超希少な多発先天奇形・重度神経発達障害症候群です。出生時から特異的な上肢の屈曲姿勢(BOS姿勢)を呈し、難治性の摂食障害・周期性嘔吐症・高度近視などを主徴とする一方で、ウィルムス腫瘍や肝芽腫の発生リスクが上昇するという点において、系統的な腫瘍サーベイランスが生命予後を左右する疾患です。
Q. Bohring-Opitz症候群とはどのような疾患ですか?まず結論だけ知りたいです
A. ASXL1遺伝子の新生トランケーティング変異によって引き起こされる、極めて稀な多発先天奇形症候群です。特徴的なBOS姿勢・重度発達遅滞・周期性嘔吐症・高度近視を主徴とし、ウィルムス腫瘍・肝芽腫に対する8歳までの定期超音波サーベイランスが管理の中核となります。
- ➤疾患の定義 → OMIM #605039・Orphanet ORPHA:97297・推定有病率100万人に1人未満
- ➤分子メカニズム → ASXL1変異→DNA脱メチル化→Wntシグナル過剰活性化という三段階の病態カスケード
- ➤主な症状 → BOS姿勢・重度発達遅滞・周期性嘔吐症・高度近視(最大-16ジオプトリ)・先天性心疾患
- ➤腫瘍リスク → 腎臓腫瘍(ウィルムス腫瘍)発生率約7%・出生後8歳まで3〜4ヶ月ごとに腹部エコー必須
- ➤遺伝カウンセリング → ほぼ全例がde novo変異・生殖細胞系列モザイクによる再発リスク約1%に留意
1. Bohring-Opitz症候群とは:疾患の定義と歴史的背景
Bohring-Opitz症候群(BOS:OMIM #605039、Orphanet ORPHA:97297)は、重度の子宮内発育遅延(IUGR)、極めて特徴的な頭蓋顔面形態、重篤かつ難治性の摂食障害、そして特異的な上肢の屈曲姿勢(BOS姿勢)を主徴とする、常染色体顕性(優性)遺伝形式をとる超希少な多発先天奇形症候群です。その推定有病率は100万人に1人未満とされており、世界の医学文献で詳細な臨床報告が行われている症例数は現在約46例にとどまっています。
💡 用語解説:常染色体顕性遺伝(じょうせんしょくたいけんせいいでん)
「常染色体」とは性染色体(X・Y)以外の46本のうちの染色体のこと。「顕性(優性)」とは、2本ある染色体の片方に変異があるだけで症状が現れることを指します。BOSは表現型が極めて重篤なため、患者本人が子を持った例は医学文献上に存在せず、臨床で診断される発症例のほぼすべてが新生突然変異(de novo変異)によるものです。
歴史的な背景を辿ると、BOSに合致する症例は1975年にF. OberklaidとD.M. Danksによって初めて文献上に記載され、当時は「Oberklaid-Danks症候群」と呼称されていました。その後1999〜2006年にかけて、ドイツの臨床遺伝学者Axel Bohringと米国のJohn Opitzらが独立して複数の症例を詳細に報告・体系化したことで、現在の病名が国際的に定着しています。また過去には「Opitz三角頭蓋様症候群」あるいは「C様症候群(C-like syndrome)」として分類されていた時期もありました。
診断技術の転換点となったのは2011年の次世代シーケンシング(エクソーム解析)の導入です。この技術によって、BOSの大部分が第20染色体長腕(20q11.21)に位置するASXL1遺伝子の新生変異に起因することが特定されました。この発見によりBOSは「形態異常の集合体」という概念から「クロマチン制御機構の破綻を基盤とするエピジェネティック起因性疾患」という新たなパラダイムへと引き上げられ、特異的な腫瘍発生リスクの解明にも繋がりました。
💡 用語解説:エピジェネティクス(epigenetics)
DNA塩基配列そのものを変えずに、遺伝子の「読まれ方(発現)」を制御する仕組みの総称です。DNAのメチル化やヒストン(DNAが巻きつくタンパク質)の修飾が代表例。エピジェネティックな制御が乱れると、胎児発生の複数のプロセスが同時に異常をきたし、多臓器にまたがる先天奇形症候群が生じることがあります。
疫学と生命予後
BOSの自然歴において最も留意すべきは、小児期初期における極めて高い死亡率です。文献による集計データでは、診断された小児の約40%が6歳に達する前に死亡しており、全報告例の26%(43例中11例)が1歳未満の乳児期に亡くなっています。乳幼児期の主な死因は複雑な先天性心疾患の悪化、喉頭軟化症・気道狭窄に起因する閉塞性睡眠時無呼吸等の呼吸停止、および反復性肺感染症です。
一方で、臨床経過には明確な個体差が存在し、重篤な合併症に対する高度な医療的介入を乗り越えて青年期・成人期早期まで生存している症例も報告されています(過去の文献では5例が成人期に達したとされます)。生存例では、生命を脅かす呼吸器・消化器症状が成長とともに段階的に安定化する傾向があります。
📊 BOSの生命予後:年齢別累積死亡リスク(過去文献データ)
26%
約40%
報告5例
出典:文献集計データ(43例)。集学的医療介入の発展により予後は改善傾向にあります。
2. 原因遺伝子ASXL1とエピジェネティック病態メカニズム
Bohring-Opitz症候群の病態の核心は、ヒストン修飾とクロマチンリモデリングに深く関与するASXL1(Additional sex combs-like 1)遺伝子の機能不全にあります。この分子メカニズムは、単一の遺伝子変異がいかにして全身多臓器にわたる形態形成の破綻を引き起こすかという、発生生物学上の重要な知見を提供しています。
💡 用語解説:ASXL1遺伝子とは
ASXL1(Additional Sex Combs Like 1)は、第20染色体長腕(20q11.21)に位置する遺伝子で、初期胚の発生過程において必須の役割を果たすエピジェネティック制御タンパク質をコードしています。ショウジョウバエの「Additional sex combs(Asx)」遺伝子のヒトにおける相同遺伝子(ホモログ)であり、ポリコーム群複合体(PcG)およびトリソラックス群複合体(TrxG)と相互作用して、発生に必要な遺伝子の転写を精緻に調整します。
💡 用語解説:ポリコーム群(PcG)複合体とは
ポリコーム群タンパク質複合体は、発生・分化に不要な遺伝子を「サイレンシング(抑制)」する役割を担うタンパク質群の総称です。一方のトリソラックス群(TrxG)は遺伝子を「活性化」方向に制御します。ASXL1はこの二つの相反する複合体の双方に関与し、遺伝子発現の精緻なオン/オフを司る”スイッチの調整役”として機能しています。
トランケーティング変異とドミナント・ネガティブ効果
BOSを引き起こす病的バリアントの大部分は、ASXL1遺伝子の後半部分(主に最後の2つのエクソン内)に生じる新生(de novo)のトランケーティング変異(機能欠失をもたらすナンセンス変異やフレームシフト変異)です。代表的な病的変異として、c.3762delT(p.N1254fs)やc.1210C>T(p.R404*)などが同定されています。
💡 用語解説:トランケーティング変異(truncating variant)
タンパク質の翻訳が途中で打ち切られ、短縮型の不完全なタンパク質が生成されてしまう変異の総称です。ナンセンス変異(終止コドンへの変化)とフレームシフト変異(挿入・欠失による読み枠のずれ)が代表的です。BOSでは、生成された短縮型ASXL1タンパク質が正常タンパク質の機能を積極的に妨害する「ドミナント・ネガティブ効果」またはハプロ不全を引き起こし、クロマチン制御ネットワークを破綻させます。
DNA脱メチル化とWntシグナル過剰活性化
患者由来の初代培養線維芽細胞を用いたマルチオミクス解析(RNAシーケンシング+DNAメチル化解析の統合)により、BOSの分子的病態の深層が近年解明されています。ASXL1の短縮型変異は、組織横断的にエピジェネノムの多層的な構造を広範に破壊することが示されています。
具体的には、BOS患者の細胞では遺伝子プロモーター領域における内在性のDNA CpGメチル化レベルの異常な低下(脱メチル化)が広範に観察されます。このDNA脱メチル化はクロマチンの異常な「開状態(アクセシビリティの上昇)」をもたらし、本来は発生の特定段階で厳密に抑制されるべき発生・形態形成経路の遺伝子群が異所性または時期外れに活性化されます。
💡 用語解説:Wntシグナル伝達経路とは
Wnt(ウィント)シグナル伝達経路は、胚発生・細胞増殖・組織の極性形成において中心的な役割を担う細胞内情報伝達システムです。カノニカル(β-カテニン依存性)経路は細胞の増殖・分化を制御し、非カノニカル(VANGL2を介する平面細胞極性:PCP)経路は細胞の移動・組織の三次元パターニングを指示します。BOSではこの両方の経路が同時に過剰活性化しており、頭蓋顔面奇形・神経発達障害・骨格形成異常の根本的な原因となっています。
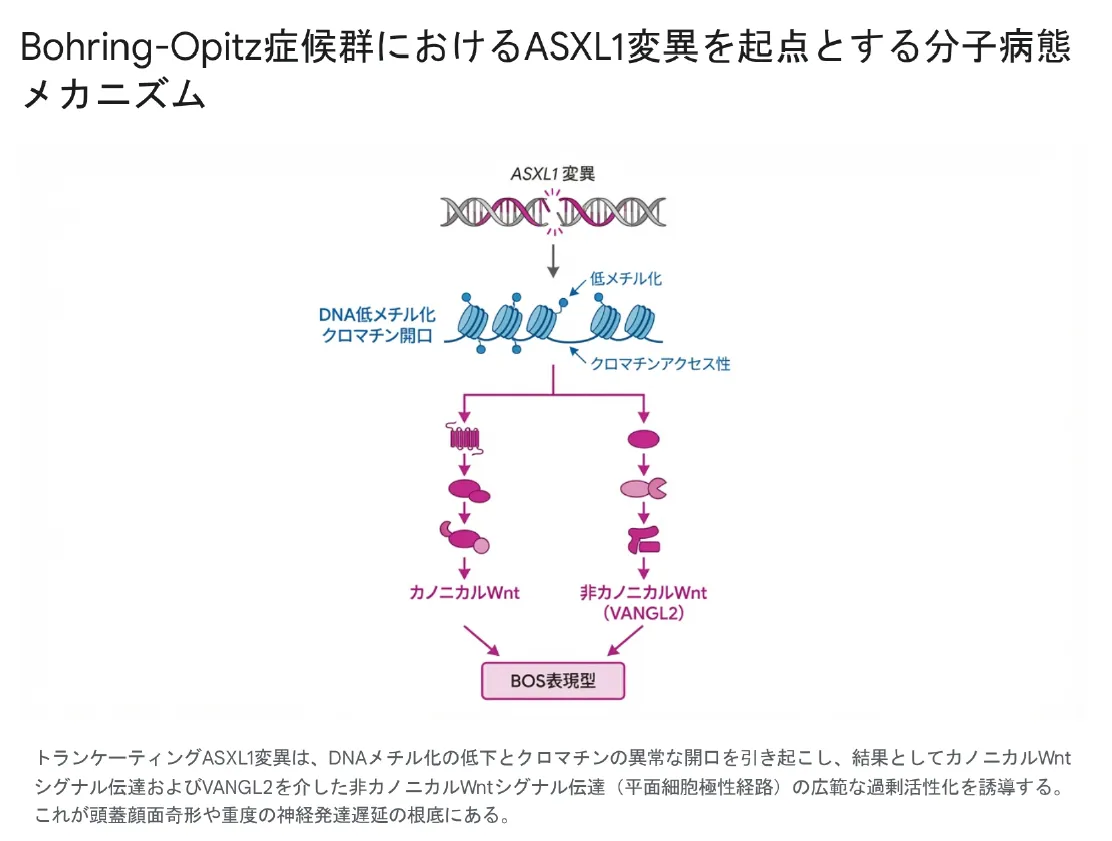
トランケーティングASXL1変異はDNAメチル化の低下とクロマチンの異常な開口を引き起こし、結果としてカノニカルWntシグナル伝達およびVANGL2を介した非カノニカルWntシグナル伝達(平面細胞極性経路)の広範な過剰活性化を誘導する。これが頭蓋顔面奇形や重度の神経発達遅延の根底にある。
体細胞変異としてのASXL1:血液がんとの関連
BOSの病態を理解するうえで特筆すべき重要な事実があります。先天性BOSを引き起こす生殖細胞系列の短縮型変異と「全く同一の分子構造」を持つ変異が、後天性の血液がん(骨髄系腫瘍)においても強力な体細胞ドライバー変異として高頻度に観察されます。
📊 ASXL1体細胞変異の検出頻度(骨髄系腫瘍別)
約45%
約30%
約16%
約10%
約6.5%
CMML:慢性骨髄単球性白血病、MDS:骨髄異形成症候群、MPN:骨髄増殖性腫瘍、AML:急性骨髄性白血病
興味深いことに、生殖細胞系列にASXL1変異を保有するBOS患児においては、この遺伝子が強力な白血病ドライバーであるにもかかわらず、小児期に骨髄系腫瘍(白血病)を発症するリスクは顕著には上昇しないと考えられています。代わりに、ウィルムス腫瘍(腎芽腫)や肝芽腫といった特定の固形腫瘍のリスクが特異的に上昇します(詳細は「腫瘍サーベイランス」の章で解説します)。

3. 主な症状と表現型スペクトラム
Bohring-Opitz症候群の臨床像は多岐にわたり、複数の臓器系に深刻な異常をもたらします。表現型の多くは成長のプロセスにおいて特異的な変容(進化あるいは退縮)を示すことが特徴です。
特異的な上肢姿勢(BOS姿勢)
本疾患を臨床的に最も強く疑わせる病的決定徴候(pathognomonic sign)のひとつが、出生時から普遍的に観察される独特の上肢姿勢「BOS姿勢」です。この姿勢は以下の解剖学的要素から構成されます。
💪 BOS姿勢の構成要素
- 両側肩関節の顕著な内旋
- 両側肘関節の持続的な屈曲
- 手関節・指関節の強い尺側偏位と屈曲
🔍 診断上の重要な鑑別点
この姿勢は関節自体の器質的な拘縮ではない。指節の皮膚のしわ(phalangeal creases)は正常に形成されている。体幹の筋緊張低下と四肢末梢の筋緊張亢進が組み合わさった神経原性の静止姿勢異常です。
乳幼児期を生き延びた患者において意図的な自発運動が可能になることもありますが、運動を停止した安静時や睡眠時には再びBOS特有の屈曲・尺側偏位姿勢へと回帰する傾向があります。なお、この姿勢は神経系の発達未熟性が顕著な小児期早期に最も目立ち、加齢とともに徐々に軽減することが多いとされています。
頭蓋顔面の特徴と眼科的異常
🧠 頭部・皮膚・体毛
- 重度の小頭症、または三角頭蓋
- 眉間〜前頭部・両眼瞼の広範な単純性血管腫(乳児期目立つが加齢で退色)
- 左右眉毛が中央でつながる癒合眉(synophrys)(加齢で顕著化)
- 急速に伸びる頭髪・爪、全身の多毛症(hypertrichosis)
👁️ 眼科・顔面
- 顕著な眼球突出(proptosis)と両眼隔離症
- 急速進行する高度近視(最大-16ジオプトリの報告あり)
- 網膜・視神経の構造的異常
- 平坦・幅広の鼻梁、顕著な前向き鼻孔
- 小顎症・下顎後退症、口蓋裂・高口蓋
- 低位耳介(後方回転が強い)、低い後頭部生え際
💡 用語解説:三角頭蓋(さんかくずがい / Trigonocephaly)
前頭縫合の早期癒合(または顕著な前頭隆起)により、上から見た頭部の輪郭が三角形に見える頭蓋形態です。BOSでは前頭縫合の早期癒合を必ずしも伴わない三角頭蓋が特徴的です。この形態はWntシグナルの異常による頭蓋顎顔面の形成不全を反映しています。
神経・発達的側面
脳のMRI検査では、脳梁の形成不全(低形成)・菲薄化、中等度の脳室拡大、前頭葉クモ膜下腔の顕著な拡大といった脳構造の異常所見が報告されています。臨床的な発達遅滞と知的障害は事実上すべての患者において重度から最重度(severe-to-profound)の範囲に分類されます。
言語機能については、表出性言語(意味のある発語)の獲得は最小限にとどまるか、生涯にわたって全く獲得されないことが多いです。しかしながら、極めて限定的な知的能力にもかかわらず、「対話的で、幸福感に満ち、好奇心旺盛(interactive, happy, and curious)」であると記述されることが多く、これは本疾患に特有の行動表現型として認識されつつあります。てんかん発作は乳児期に高頻度に見られますが、標準的な抗てんかん薬による薬物治療に対して比較的良好な反応を示し、コントロール可能なケースが多いとされています。
消化器・栄養学的合併症
多くの患児において、子宮内からの重篤な発育遅延(IUGR)に引き続いて、出生後の著しい体重増加不良(failure to thrive)が認められます。その背景には以下の消化器・摂食障害が普遍的に存在しています。
💡 用語解説:周期性嘔吐症(Cyclic Vomiting Syndrome)
激しい嘔吐が周期的に繰り返して発作的に起こる状態。BOSではこれが乳幼児期の急激な全身状態悪化・脱水・電解質異常・重度栄養不良に直結する最大の要因となります。成長・加齢に伴って徐々に改善する傾向があることが報告されていますが、乳幼児期においては生命を脅かす最重要の合併症のひとつです。
難治性の胃食道逆流症(GERD)、不顕性誤嚥(サイレント・アスピレーション)、腸管運動機能の低下も頻発し、これらが慢性的な肺損傷を引き起こして致命的な転帰をたどる引き金となります。
呼吸器・循環器系およびその他の全身異常
🫁 呼吸器系
- 喉頭軟化症・声門下狭窄による重度閉塞性睡眠時無呼吸(OSA)
- 脳幹機能未熟性に基づく中枢性無呼吸
- 睡眠障害(昼夜逆転を伴う不規則な睡眠・覚醒サイクル)
🫀 循環器系
- 一過性の徐脈・心肺停止に近い無呼吸エピソード
- 心房中隔欠損症(ASD)・肺動脈弁狭窄症・肺高血圧症・心室肥大
- 外科的修復を要する先天性心疾患合併あり
🦴 整形外科・その他
- 股関節形成不全・橈骨頭の先天性脱臼
- 重度の脊柱側弯症・後弯症
- 反復性肺炎・尿路感染症(免疫不全でなく二次性)
💡 用語解説:喉頭軟化症(こうとうなんかしょう / Laryngomalacia)
喉頭(声帯を含む気道の入り口)の軟骨が軟弱なため、呼吸時に喉頭の組織が気道内に引き込まれ、気道が狭窄する状態です。吸気時のゼコゼコ・ヒューヒューという喘鳴が特徴的で、重症例では生命を脅かす閉塞性睡眠時無呼吸を引き起こします。BOSでは声門上形成術(supraglottoplasty)や、最重症例では気管切開が必要となることがあります。
4. 鑑別診断と関連疾患スペクトラム
BOSの臨床像は他の先天奇形・知的障害症候群と部分的な重複を示すため、臨床的および遺伝学的な鑑別診断が極めて重要です。特に近年の研究では、ASXL1と関連するASXL遺伝子ファミリーの機能的ネットワークに基づき「ASXL関連障害(ASXL-related disorders)」という新たな疾患スペクトラムが定義されています。
| 疾患名 | 原因遺伝子 | BOSと共通する主な特徴 | BOSとの決定的な鑑別点 |
|---|---|---|---|
| Bohring-Opitz症候群 | ASXL1 / 常染色体顕性(De novo) | 眉間血管腫・重度発達遅延・摂食障害・筋緊張低下・小頭/三角頭蓋 | 指標:BOS姿勢・周期性嘔吐症・高度近視・出生後成長不良・腫瘍リスク↑ |
| Shashi-Pena症候群 | ASXL2 / 常染色体顕性(De novo) | 眉間血管腫・眼球突出・両眼隔離症・乳児期摂食障害・多毛・筋緊張低下 | 小頭症でなく大頭症を呈する。成長遅延なし。BOS姿勢・周期性嘔吐・高度近視なし |
| Bainbridge-Ropers症候群 | ASXL3 / 常染色体顕性(De novo) | 重度知的障害・摂食障害・多発先天奇形 | マルファン様体型(長身痩躯)・漏斗胸などの骨格異常。BOS姿勢・特有の顔貌なし |
| KLHL7関連BOS様症候群 | KLHL7 / 常染色体潜性(劣性) | IUGR・小頭症・重度摂食障害・関節拘縮・知的障害・BOS様上肢姿勢 | 典型的な癒合眉・三角頭蓋・高度近視・周期性嘔吐症が欠如。劣性遺伝形式 |
| C症候群(Opitz trigonocephaly症候群) | 多様(染色体異常等) | 三角頭蓋・特異的顔貌・重度発達遅滞・多発奇形 | 眉間血管腫・BOS姿勢・高度近視・周期性嘔吐を伴わない |
また、顔面の形態的特徴や多発奇形のパターンから、初期にはCornelia de Lange症候群(NIPBL遺伝子などのコヒーシン複合体異常)やCHARGE症候群(CHD7遺伝子異常)が疑われるケースもありますが、四肢の特異的な奇形パターンの違いや分子遺伝学的検査によって明確に鑑別されます。
5. 診断アプローチと遺伝子検査
BOSの診断は長らく、Hastingsら(2011年)が提唱した詳細な臨床診断基準に基づき、身体所見の評価から行われてきました。この基準には小頭症・三角頭蓋・顔面血管腫・特有のBOS姿勢・重度摂食障害・IUGR・重度発達遅延などが含まれます。しかし現在では、臨床的疑いに基づいて分子遺伝学的検査による確定診断を行うことが標準的アプローチとなっています。
💡 用語解説:全エクソームシーケンス(WES)・全ゲノムシーケンス(WGS)
WES(Whole Exome Sequencing)はタンパク質をコードするエクソン全体を網羅的に解析する次世代シーケンス手法で、WGS(Whole Genome Sequencing)はゲノム全体を対象とします。BOSのように表現型が非典型的であったり鑑別すべき疾患が多岐にわたる場合、あるいは単一遺伝子検査で変異が見つからない場合に選択されます。これらの検査は遺伝子検査の専門施設で実施されます。
遺伝子検査のステップ
表現型からBOSが極めて強く疑われる場合には、ASXL1遺伝子を標的とした単一遺伝子のシーケンス解析(必要に応じて欠失・重複解析を追加)が行われます。一方で、臨床像が非典型的な場合や鑑別疾患が多岐にわたる場合には、多遺伝子パネル検査またはWES/WGSが選択されます。
BOSの表現型には多様性が存在し、ASXL1変異を有していても典型的な三角頭蓋・顔面血管腫・高度近視・BOS姿勢を伴わない非典型例も報告されています(例:c.1210C>T変異の症例)。ゲノム解析結果の正確な解釈(バリアントの病原性評価)には、遺伝学的データと詳細な表現型評価(精緻なフェノタイピング)を照らし合わせる臨床遺伝専門医の高度な専門知識が不可欠です。
💡 用語解説:de novo変異(新生変異)
両親の生殖細胞(精子・卵子)または受精直後に新たに生じた変異で、両親の遺伝子には存在しない変異のことです。BOSのほぼすべての発症例がこのde novo変異によるものです。「両親が健康だから遺伝子疾患ではないはず」という誤解が診断を遅らせることがあるため、注意が必要です。ただし生殖細胞系列モザイク(後述)による例外も存在します。
6. 治療・集学的長期管理
Bohring-Opitz症候群に対する根治的な遺伝子治療や特異的治療薬は現在のところ存在しません。臨床管理の主体は、多臓器にわたる複雑な合併症の予防・治療と対症療法を包括的に行う多職種連携チーム(Multidisciplinary team)による集学的アプローチです。
消化器および栄養管理
生命維持の観点から、重度の摂食障害と周期性嘔吐による致死的な成長不良および誤嚥を回避することが最優先事項です。乳幼児期には早期からの経管栄養(経鼻胃管)の導入が必須となることが多く、胃食道逆流や不顕性誤嚥が内科的に制御不可能な重症例では胃瘻(G-tube)または胃空腸瘻(GJ-tube)の外科的造設が推奨されます。深刻な胃排出遅延を伴う場合は胃底部噴門形成術(Nissen fundoplication)などの外科的介入が適応となります。
BOSに特徴的な周期性嘔吐症に対しては、日常的な予防・維持療法としてシプロヘプタジン(抗ヒスタミン/抗セロトニン作用を持つ食欲増進薬)の定期投与が行われます。また、発作の初期兆候が見られた際にはロラゼパムやオンダンセトロン(強力な制吐薬)を速やかに投与することで症状の劇症化を防ぐアプローチが有効とされています。
呼吸器・循環器管理
睡眠時無呼吸・致死的気道閉塞の予防のため、頭蓋顔面チームや耳鼻咽喉科の早期介入が推奨されます。軽度〜中等度の閉塞性睡眠時無呼吸にはCPAP/BiPAPによる非侵襲的陽圧換気療法が用いられます。喉頭軟化症・声門下狭窄が顕著な症例では声門上形成術(supraglottoplasty)、小顎症による舌根沈下が著明な症例では下顎骨延長術(mandibular distraction)が必要となる場合があります。最重度の症例では気管切開(tracheostomy)が選択されます。循環器系については、全診断例において小児循環器科による早期の心エコー評価と定期的な心電図モニタリングが必須のプロトコルです。
発達・リハビリテーション支援
重度の知的障害および運動発達遅延に対する早期からの介入が不可欠です。理学療法士(PT)・作業療法士(OT)・言語聴覚士(ST)による包括的かつ持続的な療育支援プログラムを導入し、患児の潜在的な能力を最大限に引き出すことが求められます。てんかん発作には定期的な脳波(EEG)モニタリングに基づく標準的な抗てんかん薬による管理が行われます。
生後早期からの高度近視や視神経異常による視覚情報入力の遮断は認知発達をさらに阻害する大きな要因となるため、定期的な眼科評価と適切な視力矯正(眼鏡等)の早期導入は、単なる眼科的治療を超えた発達支援の観点から極めて重要です。就学期に達した患児に対しては、特別支援教育に関する包括的な評価と適切な教育的配慮が必要です。
7. 腫瘍サーベイランス:ウィルムス腫瘍・肝芽腫への対応
BOSの臨床管理において近年最も強く強調されているのが、特異的な小児がんの発生に対する体系的なスクリーニングプロトコルの導入です。ASXL1生殖細胞系列変異を持つBOS患者においては、ウィルムス腫瘍(腎芽腫)および肝芽腫の発生リスクが一般集団と比較して有意に上昇しており、腎臓腫瘍の発生率は約7%と推定されています。
💡 用語解説:ウィルムス腫瘍(腎芽腫 / Wilms Tumor)
腎臓の発生異常に起因する小児に特有の腎悪性腫瘍(腎芽腫)です。未分化な腎臓の細胞から発生し、主に5歳未満の幼児に発症します。早期発見・早期治療(外科的切除+化学療法)により治癒率は90%以上と高く、定期的なスクリーニングが生命予後を大幅に改善します。BOSに加えて、Beckwith-Wiedemann症候群・WAGR症候群などでもリスクが上昇することが知られています。
⚠️ 国際標準プロトコル:BOS患者への腫瘍サーベイランス
小児腫瘍学の専門家はBOS患者に対して、同様の腫瘍リスクを抱えるBeckwith-Wiedemann症候群のスクリーニングプロトコルをモデルとした厳格なサーベイランス体制を強く推奨しています。
📋 実施内容
臨床的または遺伝学的にBOSと診断されたすべての患児に対して、
「出生後直ちから8歳に達するまでの間、3〜4ヶ月ごとに定期的な腹部超音波検査」を継続的に実施することが、現在の国際的な標準医療プロトコルとして確立されています。
このサーベイランスにより、腫瘍が無症状の初期段階で早期発見され、外科的切除や化学療法による治癒率を最大化することが可能になります。BOSが疑われる・または確定している場合には、小児腫瘍科への紹介とサーベイランスプロトコルの即時開始を強く検討してください。
8. 遺伝カウンセリング:再発リスクと出生前診断
BOSのほぼすべてがde novo変異によるものであることから、遺伝歴のない家系から突発的に発生します。しかし、遺伝カウンセリングにおいて極めて重要な例外的事象として生殖細胞系列モザイク(Germline mosaicism)が報告されています。
💡 用語解説:生殖細胞系列モザイク(Germline mosaicism)
臨床的には全く無症状で健康な親の、卵子や精子といった一部の生殖細胞のみに病的バリアントが局所的に存在する状態です。体細胞(全身のほとんどの細胞)には変異がないため通常の血液検査では検出されません。この現象はCHARGE症候群やCornelia de Lange症候群などの他の古典的なde novo顕性遺伝疾患でも広く認知されているメカニズムです。
この事実が示す臨床的意義は重大です。全く健常な両親からBOSの子どもが生まれた場合であっても、「完全に偶然のde novo変異である」と断定することはできず、次回妊娠時における同胞(兄弟姉妹)への再発リスクは理論上ゼロではありません。再発リスクは約1%程度の「低いがゼロではない(low but non-negative)」リスクとして家族に適切に説明される必要があります。
すでに家系内で病的なASXL1変異が特定されている場合には、このリスクを考慮した上で、次回妊娠に対する分子遺伝学的な出生前診断(絨毛検査・羊水検査)や着床前診断のオプションが臨床的選択肢として提供されるべきです。
9. 臨床遺伝専門医からのメッセージ
よくある質問(FAQ)
🏥 遺伝子疾患・出生前診断のご相談はミネルバクリニックへ
Bohring-Opitz症候群をはじめとする希少遺伝性疾患・出生前診断に関するご相談は、
臨床遺伝専門医が在籍するミネルバクリニックへお気軽にお問い合わせください。
関連記事
参考文献
- [1] Orphanet. Bohring-Opitz syndrome. ORPHA:97297. [Orphanet]
- [2] Williamson KA, et al. Multiomics of Bohring-Opitz syndrome truncating ASXL1 mutations identify canonical and noncanonical Wnt signaling dysregulation. JCI Insight. 2023. [PMC10322691]
- [3] Russell B, et al. Bohring-Opitz Syndrome. GeneReviews®. NCBI Bookshelf. [GeneReviews – NBK481833]
- [4] ARRE Foundation. Bohring-Opitz Syndrome (ASXL1). [ARRE Foundation]
- [5] Zhou J, et al. A de novo Variant of ASXL1 Is Associated With an Atypical Phenotype of Bohring-Opitz Syndrome: Case Report and Literature Review. Front Pediatr. 2021. [Frontiers in Pediatrics]
- [6] Bohring-Opitz Syndrome (BOS). Rarechromo.org. [Rarechromo.org]
- [7] Bohring-Opitz Syndrome Foundation. Definition – Bohring-Opitz Syndrome/ASXL1. [bohring-opitz.org]
- [8] Russell B, et al. Clinical Management of Patients with ASXL1 Mutations and Bohring-Opitz Syndrome, Emphasizing the Need for Wilms Tumor Surveillance. Mol Syndromol. 2015. [PMC4760347]
- [9] Dinwiddie DL, et al. Bohring-Opitz Syndrome Caused by an ASXL1 Mutation Inherited From a Germline Mosaic Mother. Pediatr Neurol. 2018. [PubMed 29681100]
- [10] MedlinePlus. Bohring-Opitz syndrome. [MedlinePlus]
- [11] Sharo AG, et al. Examining the neurodevelopmental and motor phenotypes of Bohring-Opitz syndrome (ASXL1) and Bainbridge-Ropers syndrome (ASXL3). Front Neurosci. 2023. [Frontiers in Neuroscience]
- [12] Wang J, et al. Bohring-Opitz syndrome caused by a novel ASXL1 mutation (c.3762delT) in an IVF baby: A case report. Medicine (Baltimore). 2022. [PMC8812699]
- [13] OMIM #605039. Bohring-Opitz Syndrome. Johns Hopkins University. [OMIM]






